 |
|
||||||||||
|
|
|||||||||||
Prions
The notion that the only way for diseases to pass from one organism to another is by genetic information has long been the cornerstone of the study of infectious and heritable diseases. Even viruses, the smallest pathogenic organisms, although not technically alive, still possess genetic information in the form of DNA or RNA that is at the core of their ability to infect the host. Until recently, this has been an undisputed postulate. A revision to this well-established theory, which is at the core of the central dogma of biochemistry, was proposed due to some newly discovered evidence. A new disease that primarily affects cattle has been found. It was named "mad cow disease" for its effect on the brain of the afflicted animals. In scientific circles the disease is better known as bovine spongiform encephalitis, or BSE. Soon similar conditions were found in other species, including humans. A known and well characterized scrapie disease in sheep was placed in the same category.
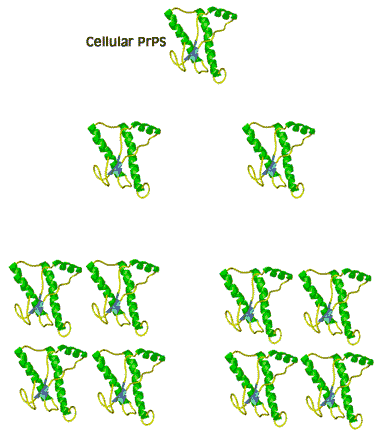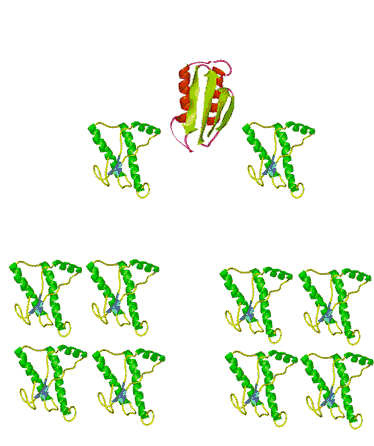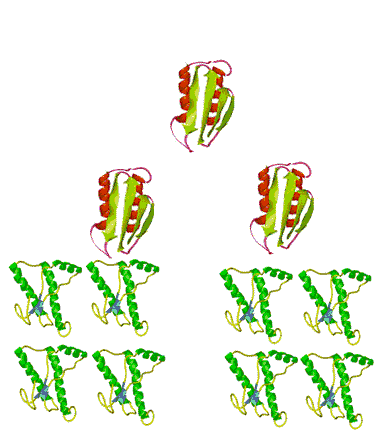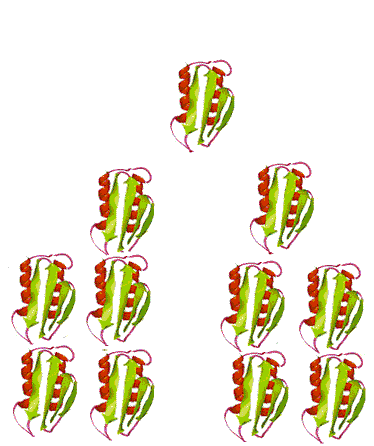
When the infectious nature of the disease was first found, it was thought that it was transmitted by a small virus. Further studies questioned that hypothesis. The disease was still transmitted, even after the infectious media was treated, by means that normally destroy genetic material, DNA, or RNA, for example by strong UV or ionizing radiation. Not even a virus can survive such a treatment. However, in this case, the disease was transmitted without hesitation. When the medium was treated by agents that only destroy proteins and leave nucleic acids intact, however, the infection was blocked. This observation immediately lead some scientists to believe that BSE is actually transmitted by proteins. Such a statement immediately raised controversy because it contradicted the central dogma of biochemistry that only change in nucleic acid composition can produce a change in protein properties. Stanley Prusiner, the winner of the Nobel Prize in Physiology/Medicine in 1997 for his discovery of prions, asserted that protein itself is capable of modifying protein content of the entire organism. His theory implied that certain proteins can come in two varieties: cellular form, designated PrPC; and infectious, misfolded form PrPSc. Later experiments confirmed that the two forms are identical in their amino acid sequences and only differ in their three-dimensional structure. According to the prion model, the disease progresses when a misfolded PrPSc molecule comes in contact with cellular form PrPC of this protein and converts them. The appearance of a misfolded molecule can be either spontaneous, or caused by feeding an animal with food containing the meat of diseased animals. Both modes of infection have exhaustive experimental confirmation. While the ‘contaminated food’ model of infection is fairly straightforward, the spontaneous appearance of the misfolded form of the prion has caused widespread controversy and attracted a host of scientists to attempt to address the problem, from kineticists to theoretical physicists. Several plausible theories have been proposed, each with its own following and its own opposition.
The problem with the spontaneous appearance of prions stems from the fact that the formation of the infectious form of the prion is thermodynamically unfavorable. If forming one molecule is unfavorable, then how can this one molecule survive in the body without reverting back into a ‘good’ cellular form, let alone convert other molecules against the ‘thermodynamic gradient’? A ‘nucleation’ theory, one of several, offers the answer to this question by suggesting that the process is very slow at first, with only a handful of molecules undergoing conversion. The misfolded molecules stabilize each other when they aggregate. After a very long time, the aggregate reaches a certain number ‘n’, at which point it is called a nucleus, thus the name for this theory. The nucleus is very stable and can quickly convert the rest of the ‘good’ molecules in their misfolded PrPSc forms. The presence of plaque-like clusters of the protein in infected cells supports this hypothesis.
The real cause for the recent increase in prion research was the discovery that animal prions, if ingested by people, can cause disorders similar to ‘mad cow disease’ in humans. In the brains of people with Creutzfeldt-Jacob disease there were found prions exactly like BSE prions. This finding announced that prions jumped the species barrier from cattle to humans. If cow prions can infect people, then it is possible that other forms of prion diseases, like scrapie in sheep, are capable of such infection. What is even more alarming is that the infectious form of the prion is more resistant to break down, so even cooked beef can contain infectious particles. Much knowledge has been gathered on prions and prion diseases in recent years, but many questions still remain to be answered. What exactly causes the debilitating neurological pathology: is it the depletion of the ‘good’ form of the prion or is it the appearance of the ‘bad’ form? What happens on the molecular level during the conversion? Do prions have some possible positive function, like they do in yeast, where prions give cells selective advantage in stressful environments? Is it possible that numerous puzzling observations in other species are caused by undiscovered prions? These and many other questions about prions will have to be answered in the years to come.
|

|
Copyright 2006, John Wiley & Sons Publishers, Inc. |