 |
|
||||||||||
|
|
|||||||||||
Shotgun Sequencing:The Shot Heard 'round the World
Heredity has been of interest to human beings for centuries, but how traits are passed to succeeding generations has only very recently come to light. While the ancient Greeks had their theories, they did not have the technology to study the mechanism of heredity. Modern genetic research can be traced back to Gregor Mendel, who in 1865 reported on hereditary patterns in pea plants. He noted that "factors" for traits such as flower color or seed shape were passed from both parents to offspring, although he did not know what this "factor" actually was. Even well into the 20th century, it wasn’t clear what molecule actually carried genetic information in the cell. Proteins, because of their complex nature, were thought for a time to be the most likely candidates. Since the discovery in the 1950s that it is DNA, written in a simple four-letter code of nucleotides, that is the hereditary material in all living organisms, sequencing, or "reading" the genetic code has become of increasing interest to scientists. While the code itself is elegantly simple in design, reading entire genomes is far from easy. The first problem is that genomes are incredibly long. Even the simplest genomes, those of viruses, are thousands of nucleotides in length. Bacterial genomes are millions of nucleotides long, and those of more complex plants and animals consist of billions of DNA base-pairs. Sequencing such large amounts of genetic information in a reasonable amount of time has only recently become possible, using a "shotgun" approach where a genome is fragmented into millions of short pieces that are sequenced simultaneously and then reassembled all at once.
In the early days of sequencing, starting in the 1970s, large amounts of DNA had to be radioactively labeled and then tediously broken into tiny pieces by chemicals. The fragments were then laboriously analyzed on large polyacrylamide gels exposed to X-ray film to determine perhaps 100 nucleotides of sequence. An improved procedure used polymerase enzymes to make radioactive DNA copies of the sequence of interest. This sped up the sequencing process somewhat, but exposure of bulky gels to large sheets of X-ray film still took days at a time, and the resulting data of perhaps 300 nucleotides was still read by the human eye and entered into computers by hand. While these old methods allowed individual genes to be sequenced with great accuracy, such manual processes were much too slow to sequence very large amounts of DNA. The interest in sequencing whole genomes necessitated a new strategy. Shotgun sequencing allows a scientist to rapidly determine the sequence of very long stretches of DNA. The key to this process is fragmenting of the genome into smaller pieces that are then sequenced side by side, rather than trying to read the entire genome in order from beginning to end. The genomic DNA is usually first divided into its individual chromosomes. Each chromosome is then randomly broken into small strands of hundreds to several thousand base pairs, usually accomplished by mechanical shearing of the purified genetic material. Each of the short DNA pieces is then inserted into a DNA vector (a viral genome), resulting in a viral particle containing "cloned" genomic DNA (Fig. 1).
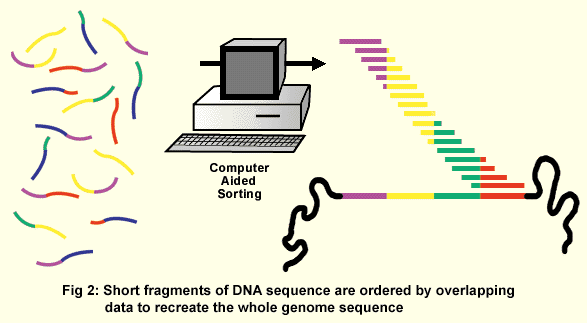
The collection of all the viral particles with all the different genomic DNA pieces is referred to as a library. Just as a library consists of a set of books that together make up all of human knowledge, a genomic library consists of a set of DNA pieces that together make up the entire genome sequence. Placing the genomic DNA within the viral genome allows bacteria infected with the virus to faithfully replicate the genomic DNA pieces. Additionally, since a little bit of known sequence is needed to start the sequencing reaction, the reaction can be primed off the known flanking viral DNA. Today’s sequencing technology is automated and much faster than the old methods. The polymerase chain reaction (PCR) is used to amplify tiny amounts of starting material with color-tagged nucleotides that are sorted in gel tubes not much thicker than a human hair. The sequences can be read automatically by a computer, with individual "runs" generating 1000 or more bases of nucleotide sequence. With this approach, banks of automated sequencing machines can churn out millions of bases of data each day. In order to read all the nucleotides of one organism, millions of individual clones are sequenced. The data is sorted by computer, which compares the sequences of all the small DNA pieces at once (in a "shotgun" approach) and places them in order by virtue of their overlapping sequences to generate the full-length sequence of the genome (Fig. 2). To statistically ensure that the whole genome sequence is acquired by this method, an amount of DNA equal to five to ten times the length of the genome must be sequenced.
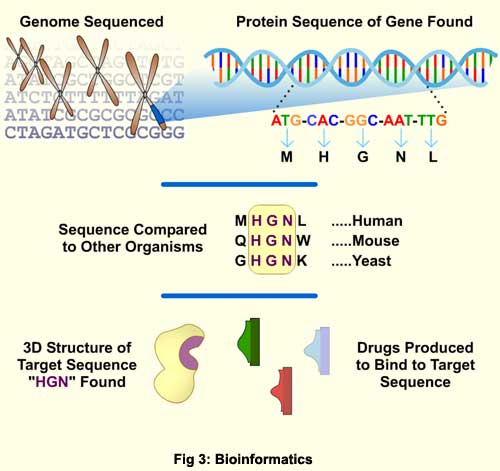
The term "shotgun" to denote sequencing of a large number of random DNA clones was first coined by F. Sanger and co-workers in 1982. They used this method to sequence the entire genome of bacteriophage l (lambda), a well studied virus that has been used as a model for how organisms regulate the use of their genes. The first free-living organism to have its genome completely sequenced was Haemophilus influenza, a pathogenic bacterium that can cause flu and meningitis. There are now many genomic sequencing projects completed and in progress, including viruses, bacteria, and fungi, as well as plants and animals. Notably, among this list is the human sequencing project , although this tremendous achievement did not come without controversy. A rivalry between two independent initiatives to sequence the human genome, the publicly funded Human Genome Project (HGP) and the private effort by Celera Genomics, came to a head in 2000 over intellectual property disputes. With both groups finishing a rough draft of the genome at about the same time, barbs flew back and forth as to who had the better data as they jostled for bragging rights. More seriously, questions arose about Celera’s specially-negotiated deal with Science magazine that would allow publication of their work without the requisite deposit of the genetic data in a public database. As a for-profit enterprise, Celera (having spent the money to do the sequencing) patented numerous novel human genes, and is currently limiting access to their data by the scientific community to protect business interests. With access to key genetic information about genetic ailments and metabolic processes on the line, the dispute raises important questions about who should have rights to human genetic data. The two groups did come to terms enough to jointly announce the completion of the draft genome at a White House ceremony in July of 2000, and agreed to simultaneously publish their data, which they did in February 2001. Far from representing the pinnacle of achievement, the sequencing of the human genome is spawning new fields of research that should prove fruitful for decades to come. Our chromosomes are believed to house about 30,000 to 40,000 genes, with each structural gene encoding a protein with a specific and important function. Even though we have their sequences, the functions of many of these genes are still unknown. Under the rubric of "bioinformatics," computer-aided biology research will mine the wealth of genetic data to try to make sense of it all. Such research is predicted to make great strides in the understanding of metabolic processes, protein function, and genetic disease. One new field is pharmacogenomics, in which information about the roles played by specific human genes in disease and drug metabolism is being used to custom design more effective and less toxic medications. It is well known in the medical community that optimal doses of certain drugs can vary greatly from person to person, and bad reactions to medications are a major cause of deaths and hospital stays. Pharmacogenomic studies are aimed at understanding how gene variations between people affect individual responses to drugs. Even a single nucleotide change, called a single-nucleotide polymorphism (SNP for short), in a gene for a critical enzyme can change the way a person reacts to a particular drug. Rapid methods (such as shotgun sequencing) for obtaining genetic data are now allowing scientists to investigate how SNPs determine individual reactions to particular medications. Another accelerating field is gene therapy, which hopes to fix medical problems by changing the genetic makeup of the patient. Certain human diseases are caused by a mutated gene that results in a once vital protein that is now missing or malfunctioning. In gene therapy, functioning copies of the wayward gene are introduced into a person’s cells to restore proper function. Identification of specific disease-causing genetic defects is the first step towards such future medical treatments. But the genes only tell part of the story, because each gene encodes a protein, and it is the proteins that actually do all the work in a cell. Bioinformatics can also be used to design drugs that will target specific proteins that cause illness (Fig 3).
Once a gene has been identified, its DNA sequence can be translated into the amino acid (protein) sequence, using the known genetic code. Comparison of the human protein to its counterpart in other organisms can reveal similarities in amino acid sequence. Such similarities can suggest which part of the protein is particularly important to its function. The sequence can also be used to predict the final three-dimensional shape of the folded protein by comparison to similar proteins whose shape is known. All this information can then be used to search for drugs that will block the protein from working. Although drug testing currently is done in traditional laboratory in vitro (test tube) experiments, searches for new drug molecules may in the future start with computer simulations, in what biologists are calling "in silico" experiments. Rather than trying them out in the lab, drugs can be initially evaluated for effectiveness and toxicity with data about protein structure and metabolic pathways stored on computer. The amount of genetic data available to scientists is growing exponentially, thanks to advanced sequencing techniques and increasing computing power. Genetic research has come far in the fifty or so years since DNA was shown to be the carrier of hereditary information. How this newfound information will be used to the benefit of mankind continues to be debated by scientists and ethicists. Nonetheless, it is becoming clear that the societal impacts made possible by rapid genome sequencing are enormous. For example, microbial genome sequences are already yielding strategies for designing drugs to combat disease-causing microbes, and microbial proteins are now used to make industrial processes more efficient and environmentally friendly. Data from the human genome is already helping us to understand more about human disease and metabolism, and shows promise in treating genetic disease and in aiding drug design. Unfortunately, detailed study of the human genome is also bringing the possibility that a person's genetic information may be used against him or her, perhaps to deny insurance coverage or employment. Genetic testing of embryos is already being used to select for desired traits in human babies. Recently, a healthy boy, a bone-marrow donor for an ailing sister, was born after the parents selectively implanted an embryo that was free of disease and also a compatible match for tissue donation. While this case was a joyous success, scenarios such as these raise disturbing questions about our ability to manipulate life. With such powerful techniques at hand, we will all have to consider how best to preserve our genetic heritage as we move into this new scientific frontier.
|
 Genomics, the field dedicated to the study of genome data, is at the heart of a revolution in genetic and medical research. With an ever increasing pace, whole genomes, the complete set of genetic instructions for an organism, are being read and recorded for creatures great and small. A growing list of viruses, bacteria, yeasts, plants and animals are joining the ranks of organisms with fully sequenced genomes. The human genetic code has almost been fully read, with the landmark publication of two separate "rough drafts" in February of 2001. The sequencing of the human genome, over 3 billion nucleotides in length, is already considered to be one of humankind’s greatest achievements, on par with the invention of the wheel in ancient times and the invention of the printing press. As those achievements changed the course of history in their time, so too the decoding of the human genome is expected to have a far-reaching impact on our society in years to come. Better medicines, disease prevention, but also perhaps genetic "typing" of individuals or selection of genetically desirable embryos all lie on the horizon of this brave new world of genomic research.
Genomics, the field dedicated to the study of genome data, is at the heart of a revolution in genetic and medical research. With an ever increasing pace, whole genomes, the complete set of genetic instructions for an organism, are being read and recorded for creatures great and small. A growing list of viruses, bacteria, yeasts, plants and animals are joining the ranks of organisms with fully sequenced genomes. The human genetic code has almost been fully read, with the landmark publication of two separate "rough drafts" in February of 2001. The sequencing of the human genome, over 3 billion nucleotides in length, is already considered to be one of humankind’s greatest achievements, on par with the invention of the wheel in ancient times and the invention of the printing press. As those achievements changed the course of history in their time, so too the decoding of the human genome is expected to have a far-reaching impact on our society in years to come. Better medicines, disease prevention, but also perhaps genetic "typing" of individuals or selection of genetically desirable embryos all lie on the horizon of this brave new world of genomic research.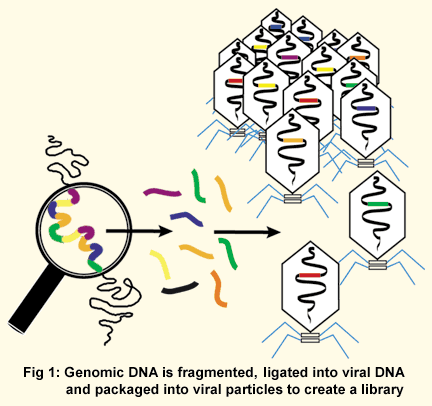
|
Copyright 2006, John Wiley & Sons Publishers, Inc. |