Introduction to Jmol Tutorial
and
Protein Secondary Structure
Introduction
Jmol is a wonderful interactive molecular display program that allows us to view proteins, nucleic acids, and many other molecules, large and small, that have been mapped into a molecular coordinate file in three dimensions. The Jmol 'applet', which fits inside a web page, is used in all the Structure Tutorials. (Jmol also exists as a stand-alone application, but is not used in that form in these tutorials.) Jmol is a free, open-source program, with a team of developers who work actively to improve it.
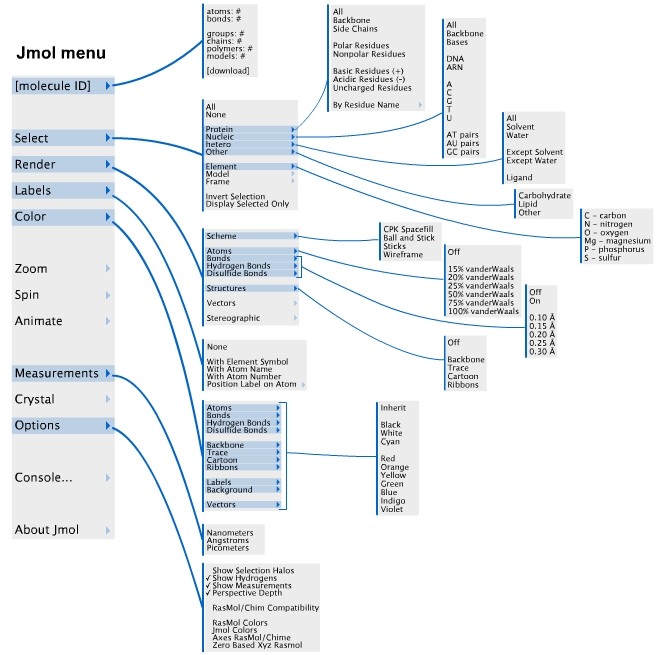
You can manipulate a molecule displayed in a Jmol applet in many ways, including rotating and zooming, using the mouse and a few keystrokes. In addition the applet has a menu, accessed by control-clicking anywhere inside the applet, or by clicking on "Jmol" in the lower right corner of the applet. The main menu and some important sub-menus are diagrammed below. The first part of this tutorial, Introduction to Jmol, will show you how to interact with the structures in Jmol using both the mouse and the Jmol menu system. The second part of the tutorial will continue the lesson, focussing on familiarizing you with the underlying level of protein folding: the elements of secondary structure.
For more information about Jmol, see the Jmol Project Home Page.
|